Contact Information
601 S Goodwin
Urbana, IL 61801
Research Interests
Research Topics
Genetics, Host-Pathogen Interactions, Regulation of Gene Expression
Disease Research Interests
Infectious Diseases
Research Description
Molecular mechanisms of Salmonella pathogenesis
Salmonella cause over one million cases of gastroenteritis and enteric fever per year in the US and lead all other foodborne bacterial pathogens as a cause of death. The long-term objectives of our research are to understand the molecular mechanisms by which Salmonella circumvents the host immune system to cause disease. SalmonellaTyphimurium provides an ideal model system to study molecular pathogenesis. The genetics of this organism are well defined and allow the simple manipulation and characterization of mutations that affect virulence. In addition, there is an excellent animal model of infection to study the effects of bacterial mutations on pathogenesis.
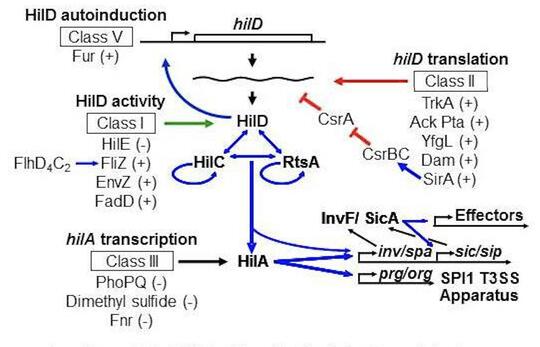
Salmonella invades the intestinal epithelium and induces inflammatory diarrhea using the Type III Secretion System (T3SS) encoded on Salmonella Pathogenicity Island 1 (SPI1). This system is transcriptionally induced in response to a variety of environmental signals such that the machinery is produced at the appropriate time and place in the intestine of the host. By performing careful genetic analyses, we have modeled the complex circuitry responsible for integrating these various environmental signals. This model is a breakthrough in Salmonella pathogenesis, explaining a plethora of published data that was often confusing. We continue to refine this model and determine the molecular mechanisms by which various environmental parameters feed into the regulator circuit. Our current foci include the roles of sRNAs in the system, a collaboration with Cari Vanderpool, and the functional characterization of the long 3’ untranslated region of the hilD mRNA.
The most serious Salmonella disease results from extraintestinal infection and bacteremia. The hallmark of these extraintestinal infections is the ability of Salmonellato survive in macrophages, which normally kill bacteria by producing a variety of antimicrobials, including superoxide (O2-). Evidence that phagocyte-produced superoxide is important in Salmonella infection is clear, but the molecular mechanism by which external superoxide kills or inhibits Salmonella is not known. Our studies have led to a paradigm shift. Contrary to current dogma, the targets of the phagocytic oxidative burst are not the bacterial DNA or other cytoplasmic molecules. Rather, we provide evidence that phagocytic superoxide kills or inhibits Salmonella by damaging an extracytoplasmic target. Our long-term goal is to understand the physiological basis of sensitivity to phagocytic superoxide and the adaptations that allow key pathogens to survive.
In an attempt to identify targets of phagocytic superoxide, we used “differential TnSeq”. We isolated large numbers of random transposon insertions in both wild type and sodCmutant backgrounds and passaged pools of these mutants through our mouse infection model. Comparing relative changes in transposon representation between wild type and sodC mutant pools recovered from the animals identifies genes that genetically interact with sodCI. We are following up on several genes identified in this screen. This includes paeA, which we believe encodes a polyamine transporter. We are studying the physiological relationship between Mg2+ and polyamines and how these factors allow adaptation to growth in the macrophage phagosome.
The tamAB locus encodes proteins that, by homology, seem to be involved in outer membrane assembly. Originally proposed as a system for assembly of a subset of beta-barrel proteins, the so-called autotransporter proteins, the actual function of TamAB is unknown. We have discovered that tamAB is a previously unrecognized member of the PhoPQ regulon and is induced when Salmonella is replicating in macrophages, suggesting that this system is important for virulence. Indeed, our preliminary data strongly support our primary hypothesis that TamAB is induced to assist Bam, the primary machine for assemble of outer membrane proteins, under the stressful conditions in the phagosome. Understanding the roles of TamAB will inform us about the conditions in the phagosome leading to outer membrane assembly stress and the mechanisms used by Salmonella to combat them. Identifying the specific proteins affected by loss of TamAB also identifies additional virulence factors critical for Salmonella pathogenesis.
Education
B.S. (Biochemistry), The Pennsylvania State University, 1984
Ph.D. (Molecular Biology), Princeton University, 1990
Postdoc. (Microbiology), Harvard Medical School, 1990-1993
Awards and Honors
University Scholar, University of Illinois, 2011-2012
School of Molecular and Cellular Biology Faculty Excellence Award, 2013
Medical Scholars Program Advisor of the Year Award, 2015
School of Molecular and Cellular Biology Excellence in Service Award, 2017
External Links
Highlighted Publications
Iwadate, Y., Golubeva, Y.A. and Slauch, J.M. (2022) Cation homeostasis: coordinate regulation of polyamine and magnesium levels in Salmonella. MBio: e0269822. https://doi.org/10.1128/mbio.02698-22
Abdulla, S.Z., Kim, K., Azam, M.S., Golubeva, Y.A., Cakar, F., Slauch, J.M., and Vanderpool, C.K. (2022) Small RNAs activate Salmonella pathogenicity island 1 by modulating mRNA stability through the hilD mRNA 3' untranslated region. J Bacteriol: e0033322. https://doi.org/10.1128/jb.00333-22
Slauch, J.M. (2022) Interplay between Rho, H-NS, spurious transcription, and Salmonella gene regulation. Proc Natl Acad Sci U S A 119: e2211222119. https://doi.org/10.1073/pnas.2211222119
Cakar, F., Golubeva, Y.A., Vanderpool, C.K. and Slauch, J.M. (2022) The small RNA MicC downregulates hilD translation to control the Salmonella pathogenicity island 1 type III secretion system in Salmonella enterica serovar Typhimurium. J Bacteriol 204: e0037821. https://doi.org/10.1128/JB.00378-21
Iwadate, Y., Ramezanifard, R., Golubeva, Y.A., Fenlon, L.A. and Slauch, J.M. (2021) PaeA (YtfL) protects from cadaverine and putrescine stress in Salmonella Typhimurium and E. coli. Mol Microbiol 115: 1379-1394. https://doi.org/10.1111/mmi.14686
Kalafatis, M. and Slauch, J.M. (2021) Long-distance effects of H-NS binding in the control of hilD expression in the Salmonella SPI1 locus. J Bacteriol 203: e0030821. https://doi.org/10.1128/JB.00308-21
Palmer, A.D. and Slauch, J.M. (2020) Envelope stress and regulation of the Salmonella pathogenicity island 1 type III secretion system. J Bacteriol 202: e00272-00220. https://doi.org/10.1128/JB.00272-20
Kim, K., Golubeva, Y.A., Vanderpool, C.K. and Slauch, J.M. (2019) Oxygen-dependent regulation of SPI1 type three secretion system by small RNAs in Salmonella enterica serovar Typhimurium. Mol Microbiol 111: 570-587. https://doi.org/10.1111/mmi.14174
Palmer, A.D., Kim, K. and Slauch, J.M. (2019) PhoP-mediated repression of the SPI1 type 3 secretion system in Salmonella enterica serovar Typhimurium. J Bacteriol 201: e00264-00219. https://doi.org/10.1128/JB.00264-19
Fenlon, L.A. and Slauch, J.M. (2017) Cytoplasmic copper detoxification in Salmonella can contribute to SodC metalation but Is dispensable during systemic infection. J Bacteriol 199: e00437-00417. https://doi.org/10.1128/JB.00437-17
Golubeva, Y.A., Ellermeier, J.R., Cott Chubiz, J.E. and Slauch, J.M. (2016) Intestinal long-chain fatty acids act as a direct signal to modulate expression of the Salmonella pathogenicity island 1 type III secretion system. MBio 7: e02170-02115. https://doi.org/10.1128/mBio.02170-15
Craig, M., Sadik, A.Y., Golubeva, Y.A., Tidhar, A. and Slauch, J.M. (2013) Twin-arginine translocation system (tat) mutants of Salmonella are attenuated due to envelope defects, not respiratory defects. Mol Microbiol 89: 887-902. https://doi.org/10.1111/mmi.12318
Golubeva, Y.A., Sadik, A.Y., Ellermeier, J.R. and Slauch, J.M. (2012) Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190: 79-90. https://doi.org/10.1534/genetics.111.132779
Slauch, J.M. (2011) How does the oxidative burst of macrophages kill bacteria? Still an open question. Mol. Microbiol 80: 580-583. https://doi.org/10.1111/j.1365-2958.2011.07612.x [doi]
Recent Publications
Abdulla, S. Z., Kim, K., Azam, M. S., Golubeva, Y. A., Cakar, F., Slauch, J. M., & Vanderpool, C. K. (2023). Small RNAs Activate Salmonella Pathogenicity Island 1 by Modulating mRNA Stability through the hilD mRNA 39 Untranslated Region. Journal of bacteriology, 205(1). https://doi.org/10.1128/jb.00333-22
Iwadate, Y., Golubeva, Y. A., & Slauch, J. M. (2023). Cation Homeostasis: Coordinate Regulation of Polyamine and Magnesium Levels in Salmonella. mBio, 14(1). https://doi.org/10.1128/mbio.02698-22
Ramezanifard, R., Golubeva, Y. A., Palmer, A. D., & Slauch, J. M. (2023). TamAB is regulated by PhoPQ and functions in outer membrane homeostasis during Salmonella pathogenesis. Journal of bacteriology, 205(10). https://doi.org/10.1128/jb.00183-23
Cakar, F., Golubeva, Y. A., Vanderpool, C. K., & Slauch, J. M. (2022). The Small RNA MicC Downregulates hilD Translation to Control the Salmonella Pathogenicity Island 1 Type III Secretion System in Salmonella enterica Serovar Typhimurium. Journal of bacteriology, 204(1), Article e00378-21. https://doi.org/10.1128/JB.00378-21
Slauch, J. M. (2022). Interplay between Rho, H-NS, spurious transcription, and Salmonella gene regulation. Proceedings of the National Academy of Sciences of the United States of America, 119(33), Article e2211222119. https://doi.org/10.1073/pnas.2211222119