
In this article, Associate Professor of Cell and Developmental Biology Stephanie Ceman discusses her research on Fragile X Syndrome.
Fragile X syndrome (FXS) is the most common form of inherited cognitive impairment, affecting 1 in 4000 males and 1 in 8000 females. FXS children often display language and motor delays as early as 9-12 months of age. In addition, 50-90% of FXS children have some symptoms and associated features of autism, including speech perseveration and other language abnormalities, gaze aversion and hand-flapping.
Individuals with FXS have mild physical abnormalities, including a larger head circumference, elongated face, large ears, and features of a mild connective tissue disorder. Fragile X syndrome is caused by the loss of expression of one gene: the Fragile X Mental Retardation 1 gene (FMR1), which is encoded on the X chromosome. We are interested in how the protein encoded by this gene, FMRP, is required for normal cognition. FMRP is expressed throughout the body; however, it is highly expressed in nerve cells (neurons) in the brain. In fact, when FMRP is absent, the part of neurons that receives input from other neurons is more immature-looking, suggesting that FMRP plays a critical role in normal neuronal function.

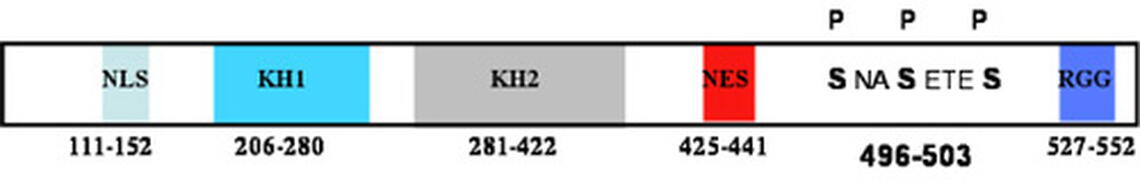
FMRP binds messenger RNAs (mRNAs). mRNAs are copies of genes that encode proteins. FMRP binds a collection of mRNAs and regulates their translation into proteins - a feature that is critical for normal neuronal function at the synapse where two neurons meet. We are specifically interested in how FMRP is regulated. The structural features of FMRP are shown in Figure 1. The RGG box binds RNA with the highest affinity. FMRP is estimated to associate with approximately 4% of brain mRNAs, and a large number of them have been identified. A central question remains: How does FMRP locate and bind its mRNAs and then regulate their translation?
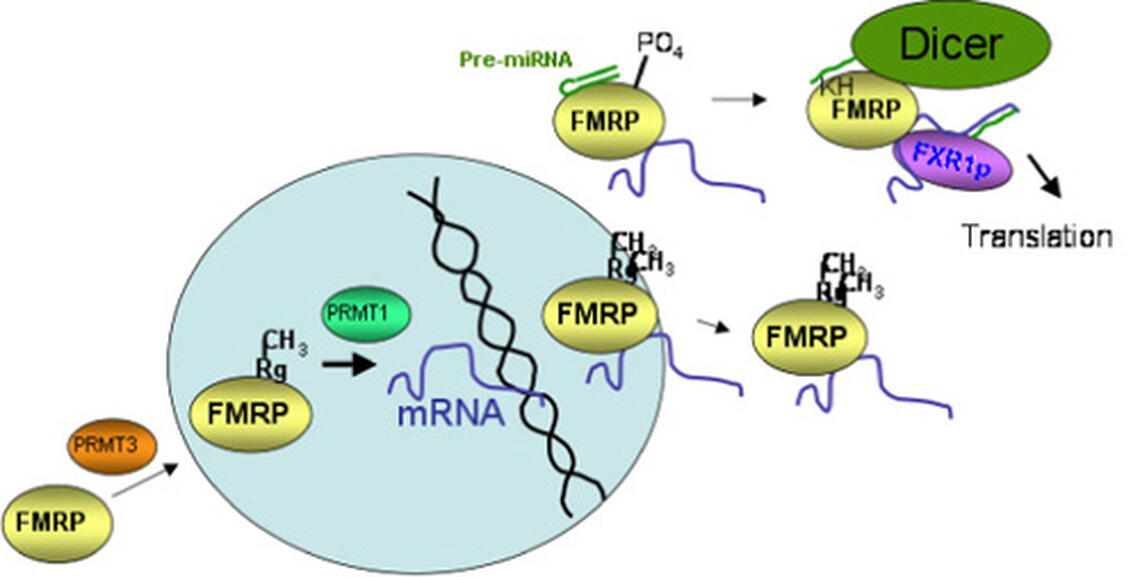
The first question we were interested in addressing was where in the cell does FMRP binds its mRNA cargoes? Cells are divided into nuclei, where all of the genomic DNA is located, and cytoplasm, where protein translation occurs. Our hypothesis was that FMRP binds its mRNA cargoes in the nucleus. We tested this using genetically engineered FMRP constructs and direct visualization of FMRP association with newly synthesized mRNAs in Xenopus laevis ooctyes in collaboration with Dr. Michel Bellini. These independent strategies allowed us to demonstrate that FMRP binds its mRNA cargoes in the nucleus, where the story of its regulatory functions begins.
We have demonstrated that the RGG box of FMRP, which is the high affinity RNA binding domain, is methylated on four of its arginines. Methylation can alter protein function by causing steric hindrance or by removing a hydrogen that participates in a hydrogen bond. We have shown that the arginines that are methylated in FMRP are important for association with mRNAs, and we are actively studying how methylation of individual arginines affects RNA binding.
Once FMRP binds its mRNAs, it regulates their translation: how it does this is also an active area of investigation in my lab. FMRP associates with the microRNA pathway. microRNAs are small RNAs that regulate the stability or translational state of the mRNAs bearing microRNA binding sites. It is now estimated that expression of nearly half of our genes is regulated by microRNAs. We have shown that FMRP is phosphorylated on three of its serines upstream of the RGG box (Fig.1). Phosphorylated FMRP associates with an increased amount of precursor microRNAs, which are unprocessed, thus, inactive microRNAs. We are currently testing the hypothesis that phosphorylated FMRP binds precursor microRNAs, sequesting them from being processed to their active state, while transporting mRNAs out to the synapse where translation is regulated (Fig.2).
This work is currently sponsored by the Spastic Paralysis Research Foundation of the Illinois-Eastern Iowa District of Kiwanis International.