
Epilepsy is a common chronic brain disorder that affects 3% of the world’s population. In epilepsy, excessive electrical activity in neurons makes them prone to misfire and trigger seizures. Nearly half of epilepsy cases are caused by genetic mutations in a variety of proteins including ion channels. Usually, seizures in epilepsy associated with genetic mutations begin from birth or childhood, thereby affecting children’s cognitive abilities and increasing their chances of developing disorders like autism or depression. Some patients with genetic epilepsy cannot be treated with existing anti-epileptic drugs, so research is needed to create new drugs to treat them.
The lab of Hee Jung Chung, Associate Professor in the Department of Molecular and Integrative Physiology, recently released a paper in Scientific Reports titled “Identifying mutation hotspots reveals pathogenetic mechanisms of KCNQ2 epileptic encephalopathy” to help fill in the gaps in our knowledge on the pathogenic mechanism underlying epilepsy-causing mutations.
“Next generation sequencing studies have increased the rate of finding new mutations linked to epilepsy. However, research on how these mutations affect protein function and eventually lead to epilepsy is lagging behind,” Chung said.
For the paper, the Chung lab studied how epilepsy mutations affect the function and neuronal expression of voltage-gated Kv7 potassium channels. Potassium ion efflux through these channels inhibits rapidly-propagating electrical signals in the neurons (called action potentials.) To open, Kv7 channels require the combination of both electrical stimuli (membrane depolarization) and the binding of a lipid called PIP2 (phosphatidylinositol 4,5-bisphosphate).
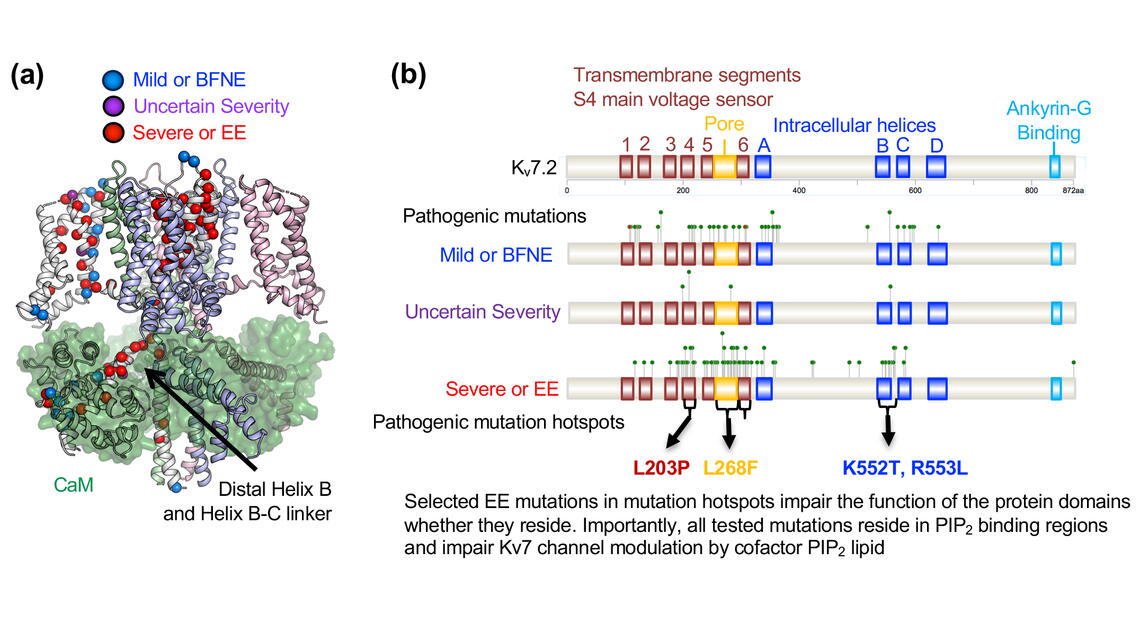
“We have more than 200 mutations in the KCNQ2 gene, which encodes the principal Kv7.2 subunit of Kv7 channels. We first wanted to know if these mutations were uniformly distributed along the channel protein sequence, or they were concentrated on specific regions of the channel.”
If the mutations are concentrated in the channel’s key functional domains, then they have a high likelihood of blocking the function of those domains. For example, mutations in the voltage-sensing domain or gate may prevent the channel from responding to membrane depolarization or opening/closing its gate. Additionally, mutations in the trafficking domain may send the channels to the “wrong address,” or send fewer channels to the “right address” in neurons.
To better understand the distribution of epilepsy mutations in Kv7.2, the Chung lab developed an statistical algorithm called “Mutation Hotspot Finder” in collaboration with Prof. Eric Bolton and his graduate student Congcong Chen in Molecular and Integrative Physiology. This algorithm allows the identification of mutation clusters in relation to their associations with functional protein domains. The resulting analysis identified four epilepsy mutation hotspots in Kv7.2. “These sites are also heavily mutated in epileptic encephalopathy, which prompt significant functional defects on Kv7 channels,” Chung explained.
The four mutational hotspots were in the transmembrane segment S4 (which is the main voltage-sensing domain in the channel), the pore loop (which mediates potassium ion permeability and selectivity), the transmembrane segment S6 (which forms the gate), and the Helix B and Helix B-C linker domains in the intracellular C-terminal tail (which binds to calcium binding protein calmodulin). Extensive experiments performed in the Chung lab revealed that selected mutations in these domains disrupted the function of the protein domains where they reside.
Additionally, the lab found that these four mutation hotspots are involved in PIP2 regulation of Kv7.2 channel. In collaboration with Profs. Emad Tajkhorshid, and Erik Procko in Biochemistry (who performed the molecular dynamics simulation and structural modeling), the lab discovered that PIP2 binds to these four hotspots. Subsequent electrophysiological studies in the Chung lab verified that selected mutations disrupt current enhancement of this channel by PIP2.
However, the PIP2 binding site discovery was mostly accidental.
Chung said, “Helix B binds to calmodulin and my lab has previously shown that Kv7.2 binding to calmodulin is critical for the channel to express properly on neuronal axons. When we found that Helix B is the mutation hotspot for severe symptomatic epileptic encephalopathy but not mild symptomatic epilepsy, we hypothesized that clinical severity of epilepsy may depend on the degree of calmodulin binding to Kv7.2. However, we didn’t see that. This led us to speculate that there must be a novel pathogenic mechanism that unifies all four mutation hotspots. Indeed, we found that PIP2 binding is disrupted by selected mutations in these hotspots.”
“Predictions are important, but predictions have to be verified with experiments. Although many epileptic encephalopathy mutations in Kv7.2 that are characterized to date are loss-of-function mutations, there also exist gain-of-function mutations. Furthermore, different mutations at the same residue are associated with diverse clinical severity of epilepsy. These are the challenges that we face in the genetic epilepsy field,” Chung said.

Overall, this paper was a group effort, incorporating the work of many undergraduate students, graduate students, and a postdoc in four different MCB labs. Specifically, Chung credits the findings in the paper to the leadership and commitment of her graduate student (Jiaren Zhang) and postdoctoral fellow (Eung Chang Kim) who performed this important study in collaborations with Profs. Bolton, Tajkhorshid, and Procko. Even though the project took over three years to complete, the lab persevered because they remembered whose lives they were trying to improve.
“We started this project because I got emails from grandparents and parents whose grandchildren and children began having neonatal seizures and had mutations on the KCNQ2 gene. They didn’t know what KCNQ2 gene does. They asked important questions such as ‘My grandkid has a mutation in KCNQ2. What is KCNQ2? How severe will his/her epilepsy be? Will his/her epilepsy be treatable?’” Chung said.
Chung stressed the importance of improving in-silico prediction tools and comprehensive experimental analyses of epilepsy variants to verify the predictions. The lab will continue this important research. Additionally, the lab is also focusing on generating knock-in mice to link the Kv7 channel dysfunction to brain circuitry and seizure generation in vivo. Understanding the pathophysiology of epileptic encephalopathy will help find better treatments for this disease.


