
A new study by researchers at the University of Illinois Urbana-Champaign suggests the overexpression of a particular gene could serve as a risk factor for Alzheimer’s disease. Their findings could also lead to better understandings of the links between Alzheimer’s disease, Fragile X Syndrome, and autism.
“The biggest challenge to us is to catch the right timing when Fmr1 participates in amyloid beta pathology,” Tsai says. “When we studied Fragile X Syndrome in the past, which is a neurodevelopmental disorder, we worked with the mice and neurons when they’re really young. So, if you monitor them closely, you will catch a time when Fmr1 is needed or dysregulated. When we study Alzheimer’s disease, which is a chronic neurological disorder, it was pretty difficult for us to catch the age or the time in mice and neurons, when Fmr1 is elevated or was provided enough, but the neurons are not degenerated yet.”
Lizarazo was successful in identifying a few points in time when Fmr1 and protein synthesis can be induced by amyloid beta peptide. The time points used by the lab were fairly early in the disease’s progression compared to other studies in the field, Tsai notes.
“I think our study may in fact bring an opportunity to reveal risk factors and therapeutic targets during the early stages of Alzheimer’s disease,” Tsai surmises. “The most important [next] step is to determine how Fmr1 may participate in cognitive decline combinations in Alzheimer’s disease.”
Now, their lab plans to test whether reducing Fmr1 can delay the progression of the disease in their animal models.
“This research could mean being able to understand a small part of the whole picture,” Lizarazo says.
A new study by researchers at the University of Illinois Urbana-Champaign suggests the overexpression of a particular gene could serve as a risk factor for Alzheimer’s disease. Their findings could also lead to better understandings of the links between Alzheimer’s disease, Fragile X Syndrome, and autism.
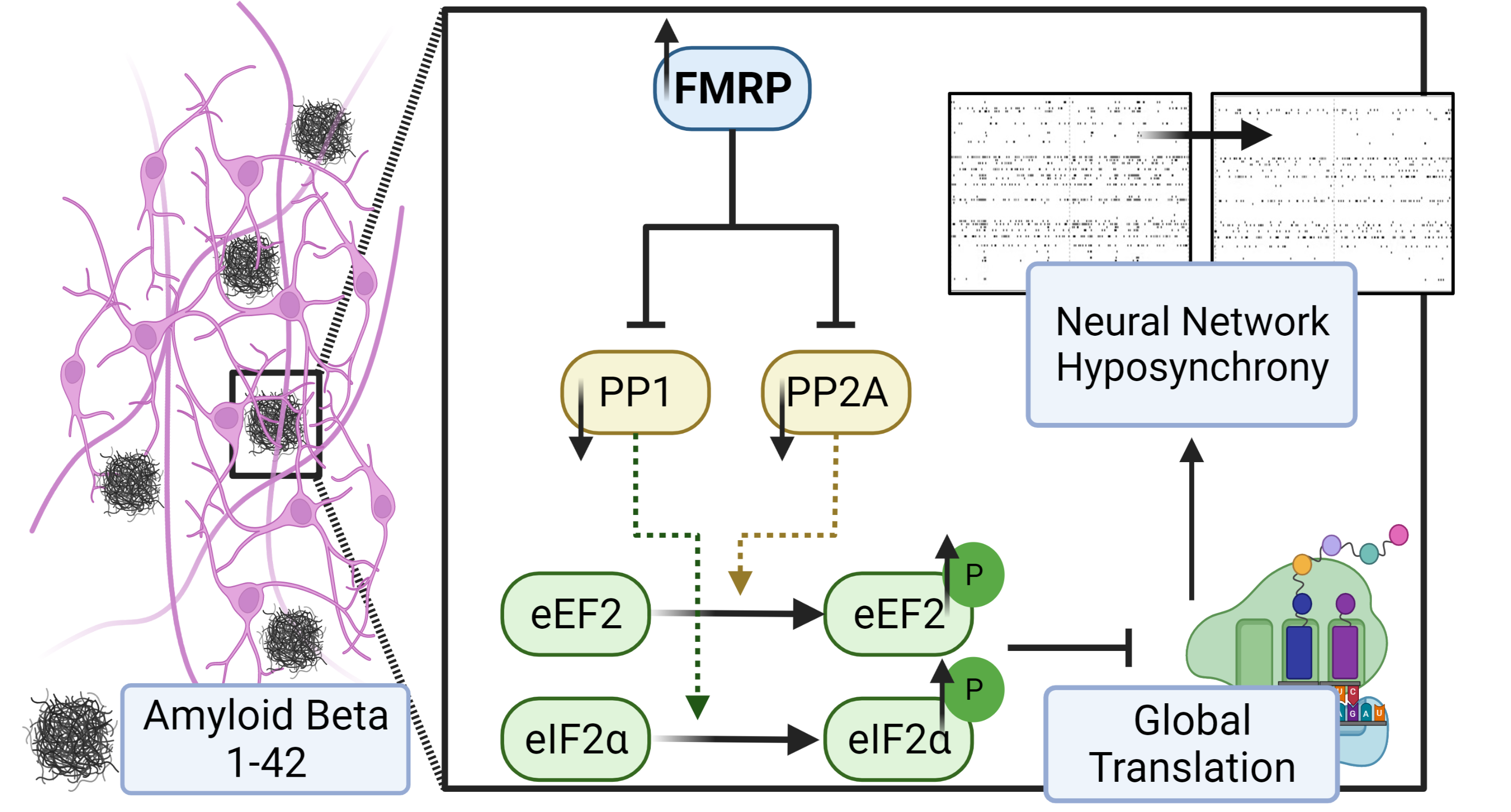
The gene at the center of the study, Fmr1, is responsible for Fragile X Syndrome, a genetic disorder that creates developmental delays and intellectual disabilities. Those who have Fragile X Syndrome do not produce the protein. However, the Tsai Lab in the School of Molecular & Cellular Biology, has found that the expression of the gene is elevated in its Alzheimer’s disease models. The results of their study were recently published in the Journal of Cellular Physiology.
Researchers believe over-accumulation of amyloid beta peptide (Aβ) in the human brain is one of the main causes of Alzheimer’s disease. According to the National Institute on Aging, the disease is the most common cause of dementia.
“In order to study how this peptide causes neurodegeneration, we applied synthetic amyloid beta peptide in our neurons [in mice] and we tried to study how the neurons responded to this peptide,” Nien-Pei Tsai, a professor of molecular and integrative physiology, explains. “That’s how we found out that the expression of Fmr1 is being elevated when the neuron sees this peptide for the first time."
Of significance, the study specifically shows that Aβ42 induces the expression of Fmr1, which represses the expression of protein phosphatase 1 and 2A. That leads to the phosphorylation of the eukaryotic translation factors and subsequent translational suppression. The Tsai Lab’s findings that phosphatase plays an important role during the progression of cellular stress could impact the development of new therapies down the road.
“Our work contributes to the understanding of a very complex process within a very complex disease,” first author Simón Lizarazo, a molecular and physiology PhD student, says. “The more insights we get about cellular stress during the progression of Alzheimer’s disease, the easier it will be for scientists to come up with a therapeutical target.”
Because of Fmr1's role in Fragile X Syndrome, their research has revealed a precarious balancing act for the protein from a person’s birth to their death. In addition to impacting both Fragile X Syndrome and Alzheimer’s disease, the absence or “silence” of Fmr1 can also lead to a genetically inherited form of autism.
“The biggest challenge to us is to catch the right timing when Fmr1 participates in amyloid beta pathology,” Tsai says. “When we studied Fragile X Syndrome in the past, which is a neurodevelopmental disorder, we worked with the mice and neurons when they’re really young. So, if you monitor them closely, you will catch a time when Fmr1 is needed or dysregulated. When we study Alzheimer’s disease, which is a chronic neurological disorder, it was pretty difficult for us to catch the age or the time in mice and neurons, when Fmr1 is elevated or was provided enough, but the neurons are not degenerated yet.”
Lizarazo was successful in identifying a few points in time when Fmr1 and protein synthesis can be induced by amyloid beta peptide. The time points used by the lab were fairly early in the disease’s progression compared to other studies in the field, Tsai notes.
“I think our study may in fact bring an opportunity to reveal risk factors and therapeutic targets during the early stages of Alzheimer’s disease,” Tsai surmises. “The most important [next] step is to determine how Fmr1 may participate in cognitive decline combinations in Alzheimer’s disease.”
Now, their lab plans to test whether reducing Fmr1 can delay the progression of the disease in their animal models.
“This research could mean being able to understand a small part of the whole picture,” Lizarazo says.

