Contact Information
Research Interests
Research Topics
DNA Biology, Genetics, Genomics, Host-Pathogen Interactions, Microbial Physiology
Research Description
Mechanisms of DNA replication stress and genome instability
We are broadly interested in the phenomenon of Genetic Death via chromosome inactivation, — with the final objective of killing undesirable cells by undermining their genetic material. Specifically, we are interested in the chromosome lesions (DNA lesions or conditions that block the chromosome cycle), — in their formation, repair (or inability to repair) and misrepair (leading to DNA rearrangements).
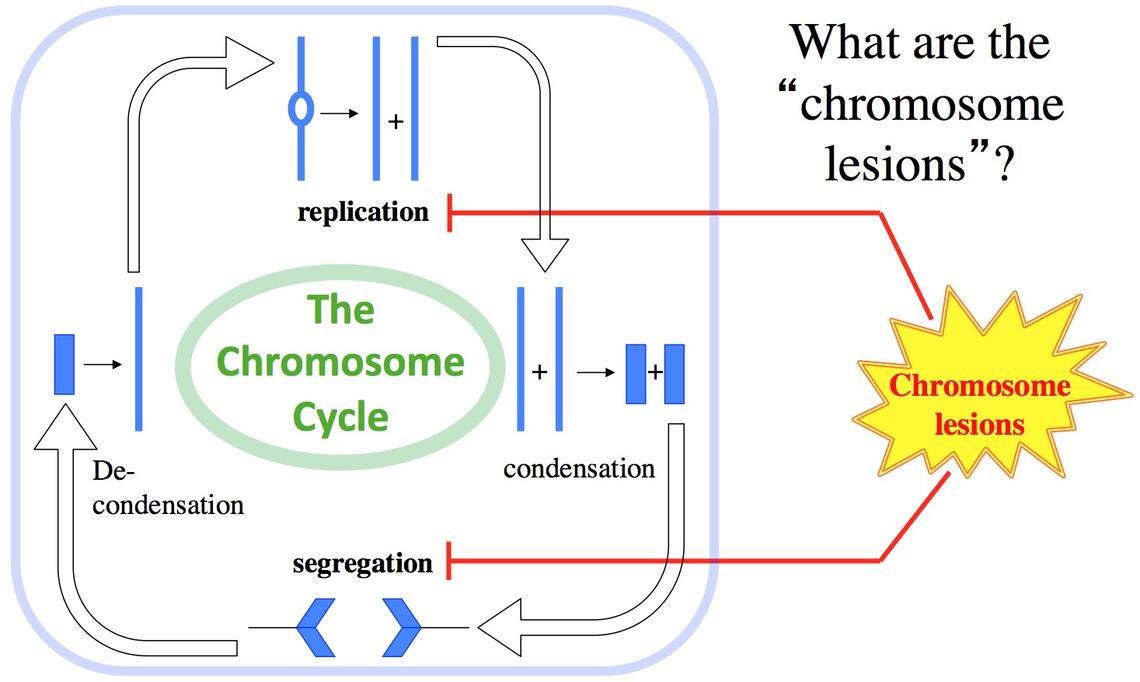
Our overarching questions about the nature of Genetic Death are as follows: — How is it possible to kill the chromosome without DNA damage? — How does DNA damage kill when the chromosome can still replicate and develops no double-strand DNA breaks? — How is it possible to trick cell into shattering their own chromosomes beyond repair?
To answer our questions, we use a powerful combination of genetic, biochemical, physical and computational approaches. We do our studies in Escherichia coli, mostly for the tractability of results, but other potentially interesting microorganisms, like Deinococcus radiodurans, are always a possibility.
Thymineless death (TLD)
The thyA mutants, unable to synthesize thymidine (dT), rapidly die without thymidine supplementation. Since the bulk of dT resides in the chromosomal DNA, TLD was always considered a chromosome replication phenomenon. Indeed, stationary cells do not replicate and are resistant to TLD, so T-starvation was naturally assumed to specifically target replication forks. Various models were proposed to explain how problems at inhibited replication forks and their eventual breakage turn deadly for the chromosome.
However, we have found that cells with demonstrably no replication in their chromosomes, but otherwise normal metabolism, are fully sensitive to TLD, indicating a very different mechanism of cell killing. Recently, we have also found that chromosome fragmentation during thymidine starvation can be completely suppressed without any influence on TLD kinetics. Thus, the two major chromosomal processes of the previous TLD models, replication and fragmentation, turn out to have no role in TLD. New TLD models need to be formulated.
We have also discovered a substantial pool of low-molecular weight (LMW) dT in the cell. A lot is still unknown about it: — The composition of this LMW-dT pool; — The main enzymes pumping free dT into it, as well as out of it; — The major processes (besides DNA replication) that tap into this LMW-dT pool. In the mutants unable to recruit free dT from this LMW-dT pool, we observe dT-hyperstarvation, reflected in the unusually fast thymineless death, which is associated with two additional striking phenomena: 1) rapid loss of the chromosomal DNA, again independently of its replication; 2) severe cytoplasm instability and/or cell envelope disintegration.

This means that, in addition to chromosomal DNA replication (and integrity, as it turns out), dT is also used to support cytoplasm or envelope integrity. We are working on figuring out whether the chromosomal problems in this mutant during dT-starvation develop independently, precede or are caused by the cytoplasm/envelope problems.
In general, identification of the considerable LMW-dT pool inside the cell revealed the second dimension of the thymineless death phenomenon, with multiple future research venues.
Oxidative DNA damage
Hydrogen peroxide (HP) is an unusual molecule in that it is inert against typical organic molecules, like proteins or nucleic acids (for example, it is stable in plastic containers for years), but at the same time kills any kind of cell on contact. Its extreme life-directed reactivity is explained by transition metals, especially iron, which are always present in cells and readily donate electrons to HP. The extra electrons split HP molecule, generating hydroxyl radicals, — the most reactive chemicals that could be made inside the cell. This is exactly how HP is used by our leukocytes to kill invading microbes.
Remarkably, the concentration of HP inside the phagosomes is four orders of magnitude lower that the killing HP concentrations we have to use in bacterial cultures. The HP concentrations that kill in cultures have to be high because bacterial cells have powerful scavenging capacities against HP. How these HP scavenging capacities are neutralized by our leukocytes remains a puzzle.
One phenomenon that partially explains the puzzling efficiency of the extremely low HP concentrations in vivo is potentiation of HP toxicity by co-treatment with other simple chemicals, like nitric oxide (NO). Leukocytes are known to pump NO into the phagosome, but the exact mechanisms of how HP+NO treatment kills bacterial cells is not known. One intriguing discovery that we have made in HP+NO-treated bacteria is the so-called "catastrophic chromosome fragmentation", when the bacterial chromosome suffers 100-1,000 double-strand DNA breaks and becomes irreparable.
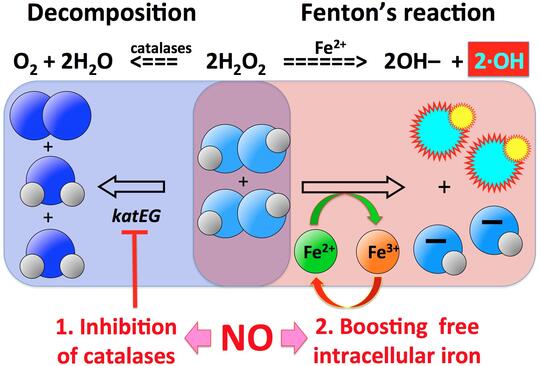
Our major questions in this project are: — What is the nature of these multiple HP+NO-induced double-strand DNA breaks? — What is the source of reduced iron to generate enough hydroxyl radicals for catastrophic chromosome fragmentation? — How much is the inhibition of HP scavenging involved in potentiation? — What is the specific role of NO in both iron recruitment and in inhibition of HP scavenging? — Are there any other treatments (simple chemicals, ambient conditions) that synergize with HP treatment?
Our current results allow us to be optimistic about eventually being able to explain the dramatic differences between the bactericidal HP concentrations in cell cultures versus phagosomes of leukocytes.
RNA-containing chromosomal lesions (R-lesions)
The main contamination of DNA turns out to be RNA, and not only single ribonucleotides occasionally misincorporated in the DNA chains, but also RNA transcripts that hybridize back with their duplex DNA templates, forming R-loops in which RNA strands have no covalent linkages to DNA strands. The cell does not tolerate either type of RNA presence in the DNA, removing RNA residues with specialized enzymes, RNases H. Every cell has a dedicated RNase H to remove single DNA-rNs, as well as (usually) another dedicated RNase H to remove R-loops. Although the two RNase H substrates are very different, and single mutants in one or the other RNase H display no or modest growth defects, the mutant lacking both enzymes is grossly inhibited. The reason for this synergy between the two RNase H defects is currently unknown. We have found that the double RNase H-deficient mutants form filamentous cells filled with DNA, whose survival depends on recombinational repair, suggesting formation of chromosomal lesions and the inability to repair some of them faithfully. The nature of these so-called R-lesions (RNA-containing chromosome lesions) is completely unknown, although it is clear that they start with formation of R-loops.
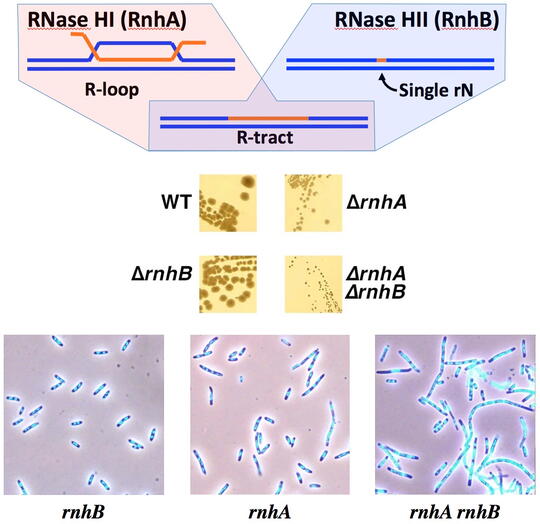
We have speculated that these chromosomal lesions could be R-tracts (DNA duplexes with extended ribonucleotide runs, incorporated in one of the strands). However, currently we are unable to detect these R-tracts in the genomic DNA of the RNase H-deficient mutants. There could be two explanations for our frustration: 1) there are too few of R-tracts per chromosome, so our detection protocols simply lack sensitivity; 2) the R-lesions are not R-tracts, but something else. We address the challenge of R-lesion detection with two complementary approaches: 1) by screening for mutants that depend on the RNase H for viability (so-called "synthetic lethals" with RNase H inactivation); 2) by finding treatments that kill RNase H-deficient mutants. In both cases, we hope to find conditions (either genetic defects or treatments) that amplify R-lesions, — which will facilitate their detection. In other words, under these conditions, we should either start detecting R-tracts or should detect something else that kills RNase H mutants, — revealing alternative R-lesions.
Replication intermediates
Inactivation of DNA ligase preserves the original size of replication intermediates in vivo (by preventing their maturation into the full-length chromosomal DNA), but excision repair breaks replication intermediates into smaller pieces. For many years, this post-replication breakage confused in vivo researchers into thinking that both the leading and the lagging strand are synthesized in similar-size pieces (Okazaki fragments). Recently, we have inactivated all known excision repair activities in the E. coli ligA mutant and found the true size of replication intermediates: they are indeed small on the lagging strand (~ 1 kb), but much longer on the leading strand (~50 kb), although still requiring periodic re-priming.

Our newly-developed ability to separate the leading strand RIs from the lagging strand RIs opens up possibilities to explore the regulation of either species, separately. For example, we are in a position to ask what determines the frequency of the periodic re-priming on the leading strand, testing such mutants or conditions as: 1) replication-blocking DNA lesions; 2) defects or overproduction of the major nucleoid-associated proteins; 3) topoisomerase defects; 4) the copy number of primase. We are also planning to inquire into what determines the cadence of primase on the lagging strand, by measuring the length of Okazaki fragments in: 1) the various primase mutants, as well as under- and over-expressing constructs; 2) mutants in single-strand DNA-binding (SSB) protein; 3) mutants in the HolC and HolD subunits of the replisome, which are considered to be responsible for managing SSB on the single-strand DNA at the replication fork. Replication intermediates in vivo may finally yield their mechanistic secrets!
Education
M.A. (Biochemistry) University of Novosibirsk, Russia, 1985
Ph.D. (Molecular Biology) Institute of Cytology and Genetics, Novosibirsk, Russia, 1990
Postdoctoral (Molecular Genetics) Institute of Molecular Biology, University of Oregon, 1991-2000
Additional Campus Affiliations
Professor, Microbiology
External Links
Highlighted Publications
Representative Publications
Our publications in the last five years:
Kuzminov A (2024) Bacterial nucleoid is a riddle wrapped in a mystery inside an enigma. J. Bacteriol. In press
Cronan GE and Kuzminov A (2024) Degron-Controlled Protein Degradation in Escherichia coli: New Approaches and Parameters. ACS Synth. Biol. In press PMID: 38317378, DOI: 10.1021/acssynbio.3c00768 (Online ahead of print)
Das S, Forrest J and Kuzminov A (2023) Synthetic lethal mutants in Escherichia coli define pathways necessary for survival with RNase H deficiency. J. Bacteriol. 205: e0028023. PMID: 37819120
Mahaseth T and Kuzminov A (2022) Catastrophic chromosome fragmentation probes the nucleoid structure and dynamics in Escherichia coli. Nucleic Acids Res. 50: 11013-11027. PMID: 36243965
Rao TVP and Kuzminov A (2022) Robust linear DNA degradation supports replication-initiation-defective mutants in Escherichia coli. G3 (Bethesda) 12:jkac228. PMID: 36165702
Khan SR and Kuzminov A (2022) Thymine-starvation-induced chromosomal fragmentation is not required for thymineless death in Escherichia coli. Mol. Microbiol., 117: 1138-1155, PMID: 35324030
Agashe P and Kuzminov A (2022) Nitric oxide precipitates catastrophic chromosome fragmentation by bolstering both hydrogen peroxide and Fe(II) Fenton reactants in E. coli. J. Biol. Chem. In press (doi: 10.1016/j.jbc.2022.101825), PMID: 35288189
Rao TVP and Kuzminov A (2022) Oxidative damage blocks thymineless death and trimethoprim poisoning in Escherichia coli. J. Bacteriol. 204: e00370-21 PMID: 34633866
Rao TVP and Kuzminov A (2021) Electron microscopy reveals unexpected cytoplasm and envelope changes during thymineless death in Escherichia coli. J. Bacteriol., 203: e0015021. PMID: 34152201
Agashe P and Kuzminov A (2021) Catalase inhibition by nitric oxide potentiates hydrogen peroxide to trigger catastrophic chromosome fragmentation in Escherichia coli. Genetics, 218: iyab057. PMID: 34027548,
Kouzminova EA and Kuzminov A (2021) Ultraviolet-induced RNA:DNA hybrids interfere with chromosomal DNA synthesis. Nucleic Acids Res. 49: 3888-3906. PMID: 33693789,
Rao TVP and Kuzminov A (2020) Exopolysaccharide defects cause hyper-thymineless death in Escherichia coli via massive loss of chromosomal DNA and cell lysis. Proc. Natl. Acad. Sci. USA, 117: 33549-33560. PMID: 33318216
Kuzminov A (2019) Half-Intercalation Stabilizes Slipped Mispairing and Explains Genome Vulnerability to Frameshift Mutagenesis by Endogenous "Molecular Bookmarks". BioEssays, 41(9):e1900062. PMID: 31379009
Khan SR and Kuzminov A (2019) Thymineless death in Escherichia coli is unaffected by the chromosomal replication complexity. J. Bacteriol. 209: e00797-18. PMID: 30745374
Rao TVP and Kuzminov A (2019) Sources of thymidine and analogs fueling futile damage-repair cycles and ss-gap accumulation during thymine starvation in Escherichia coli. DNA Repair, 75: 1-17. PMID: 30684682,
Cronan GE, Kouzminova EA and Kuzminov A (2019) Near-continuously synthesized leading strands in Escherichia coli are broken by ribonucleotide excision. Proc. Natl. Acad. Sci. USA, 116: 1251-1260. PMID: 30617079
Kuzminov A (2018) When DNA Topology Turns Deadly - RNA Polymerases Dig in Their R-Loops to Stand Their Ground: New Positive and Negative (Super)Twists in the Replication-Transcription Conflict. Trends Genet. 34: 111-120. PMID: 29179918
Recent Publications
Cronan, G. E., & Kuzminov, A. (2024). Degron-Controlled Protein Degradation in Escherichia coli: New Approaches and Parameters. ACS synthetic biology, 13(2), 669-682. https://doi.org/10.1021/acssynbio.3c00768
Kuzminov, A. (2024). Bacterial nucleoid is a riddle wrapped in a mystery inside an enigma. Journal of bacteriology, 206(3). https://doi.org/10.1128/jb.00211-23
Das, S., Forrest, J., & Kuzminov, A. (2023). Synthetic lethal mutants in Escherichia coli define pathways necessary for survival with RNase H deficiency. Journal of bacteriology, 205(10). https://doi.org/10.1128/jb.00280-23
Agashe, P., & Kuzminov, A. (2022). Nitric oxide precipitates catastrophic chromosome fragmentation by bolstering both hydrogen peroxide and Fe(II) Fenton reactants in E. coli. Journal of Biological Chemistry, 298(4), Article 101825. https://doi.org/10.1016/j.jbc.2022.101825
Khan, S. R., & Kuzminov, A. (2022). Thymine-starvation-induced chromosomal fragmentation is not required for thymineless death in Escherichia coli. Molecular Microbiology, 117(5), 1138-1155. https://doi.org/10.1111/mmi.14897